Informationen zu Krankheitsbildern
Auf dieser Seite finden sie Informationen zu ausgewählten Fehlbildungen, Krankheitsbildern und -komplexen
Morbus Hirschsprung
Der M. Hirschsprung gehört zu den seltenen Fehlbildungen. Als selten gilt eine Fehlbildung oder Erkrankung dann, wenn nicht mehr als 5:10.000 Neugeborene von der Fehlbildung betroffen sind.
Bei dieser Fehlbildung sind in einem wechselnd langen Abschnitt des Dickdarms keine Ganglienzellen angelegt. Dieser Abschnitt befindet sich stets unmittelbar oberhalb des Anus und betrifft einen unterschiedlich langen Darmanteil. Die Ganglienzellen koordinieren die Bewegung des Darms. Der von der Fehlbildung betroffene Darmanteil kann sich nicht bewegen und ist reaktiv eng gestellt.
Der Darm oberhalb dieses unbeweglichen Darmanteils versucht durch vermehrte Kraftanstrengung gegen den Widerstand des unbeweglichen Darms an zu arbeiten. Dadurch wird dieser Darm größer und kräftiger. Der Kaliberunterschied zwischen dem eng gestellten Darm und dem Darm, der gesund, aber erweitert ist, nimmt im Laufe des Lebens immer weiter zu. Beim Neugeborenen ist der Kaliberunterschied oft noch nicht so ausgeprägt.
Zum Verständnis möchten wir kurz die embryologische Entwicklung des Dickdarms erklären.
In der 4 – 10 Schwangerschaftswoche entwickelt sich der Darm des Kindes. Die Ganglienzellen migrieren (wandern) vom Mund in Richtung des Anus. Diese Migration findet in der frühen Embryonalentwicklung statt und ist zum Zeitpunkt der Geburt abgeschlossen.
Der Verlauf des Dickdarms von oral („oben) nach aboral(„unten“) ist: Zökum (Blinddarm), Kolon ascendens, Kolon transversum, Kolon descendens, Kolon sigmoideum, Rektum, Anus.
Die Migration von Ganglienzellen findet stets kontinuierlich statt. Die Ganglien migrieren dabei von oral nach aboral. Ganglienzellen können nicht „springen“.
Die Inzidenz des M. Hirschsprung (MH) wird mit 1:3500 bis 1:5000 Geburten angegeben. In Deutschland werden pro Jahr ca. 780.000 Kinder geboren, die Anzahl Neugeborener mit M. Hirschsprung beträgt daher ca. 150 - 200/Jahr.
Diagnostik und Therapie des M. Hirschsprung stellen eine Herausforderung dar, da die Fehlbildung nicht unmittelbar zu erkennen ist. Wird die Diagnose gestellt, bedarf es vorbereitender Schritte, um die Operation adäquat zu planen.
Diagnostischer Algorithmus
- Versuch, den Darm zu spülen. Gabe von Laxanzien. Anpassung der Ernährung.
- Kolonkontrasteinlauf Die Kolonkontrastdarstellung ermöglicht die Übergangszone zwischen fehlgebildetem und gesundem Darm vor der entscheidenden Operation zu lokalisieren und die Operation zu planen. Diese Untersuchung ist wichtig, da eine Fehleinschätzung des Ausmaßes der Fehlbildung verhindert werden muss. Eine Fehleinschätzung kann zum einen dazu führen, dass zu viel gesunder Darm entfernt wird. Es ist zum anderen auch möglich, dass zu wenig fehlgebildeter Darm entfernt wird und die Operation deshalb nicht erfolgreich sein kann.
- Rektumbiopsie
- Anorektale Manometrie (Druckmessung im Schließmuskel): Optional
- Alternative zur Röntgenuntersuchung: Kolonmapping: Dies ist ein zusätzlicher chirurgischer Eingriff, bei dem die Bauchhöhle eröffnet wird und zahlreiche Gewebeproben aus dem Darm entnommen werden, um so eine „Landkarte“ = Map des Dickdarms zu erstellen.
Die Operation:
Die Methode der Wahl ist der so genannte Transanale Durchzug, optional mit Laparoskopie. Zwingend erforderlich muss ein intraoperativer Schnellschnitt (Untersuchunsgtechnik des Pathologen) zur Festlegung der Resektionsgrenzen durchgeführt werden.
Bei einer Lege artis durchgeführten Operation werden die ersten 4 – 6 cm des Rektums transanal präpariert. Dann wird das Peritoneum (Bauchfell) von transanal eröffnet, indem die Muskulatur des Darms durchtrennt wird. Anschließend wird der nicht Ganglienzellen beinhaltende Darm in Ganzwand präpariert und mit wenigen Mesenterialligaturen transanal mobilisiert. Die schwierigste Phase der Operation ist die Präparation bis an das Peritoneum. Die Mobilisation des Darms in Ganzwand ist dann einfacher möglich. Die Operation endet mit der Anastomose des Ganglienzellentragenden Darmes am Anus
Die Durchführung einer Schnellschnittuntersuchung während der Durchzugsoperation ist wichtig, da eine Fehleinschätzung des Ausmaßes der Fehlbildung verhindert werden muss. Eine Fehleinschätzung kann zum einen bedeuten, dass zu viel gesunder Darm entfernt wird. Es ist zum anderen auch möglich, dass zu wenig fehlgebildeter Darm entfernt wird und die Operation deshalb nicht erfolgreich sein kann.
Durch den Schnellschnitt wird festgelegt, ob die richtige Stelle des Darms ausgewählt wurde. Werden im Schnellschnitt keine Ganglienzellen nachgewiesen, muß die Operation fortgeführt werden. Die Entfernung des Darms muss so lange fortgeführt werden, bis ein ganglienzellentragender Darmabschnitt identifiziert wird. Der Beweis, dass im Schnellschnitt Ganglienzellen zu sehen sind, ist entscheidend, da vorher die Operation nicht beendet werden darf.
Nach der Operation:
Die Kinder werden auf unserer interdisziplinärene pädiatrischen Intensivstation überwacht. Die Eltern begleiten die Kinder und wohnen mit den Kindern auf der Intensivstation. Ein besonderer Schwerpunkt liegt auf der Schmerztherapie. In unserer Klinik beginnen wir unmittelbar nach der Operation am Operationstag mit dem Kostaufbau. Nach Aheilung der Anastomose wird erneut mit den Spülungen angefangen, um den Darm an die nun veränderte Anatomei und Belastung zu gewöhnen. Ziel ist der Aufbau eines funktionierenden Kontinenzorgans.
Omphalozele & Gastroschisis
Bei beiden Krankheitsbildern handelt es sich um angeborene Fehlbildungen, bei denen ein Defekt der vorderen Bauchwand besteht. Durch diesen Defekt („Loch in der Bauchwand“) gelangt ein Teil der Bauchorgane vor die Bauchhöhle des Embryos und entwickelt sich außerhalb seines Bauches. Bei der Gastroschisis liegt der Bauchwanddefekt typischerweise rechts vom Bauchnabel und ist meist klein, einige Zentimeter im Durchmesser. Die Omphalozele beschreibt eine Nabelschnurhernie – bei ihr liegt das „Loch“ in der Bauchwand an der nicht korrekt verschlossenen Nabelschnurbasis. Es kann dabei kleinere bis teils auch größere Ausmaße annehmenund hindurch herniert ein Teil der Bauchorgane in die Nabelschnur hinein. Die Organe sind somit von einem Bruchsack = der Nabelschnur umgeben, im Gegensatz zur Gastroschisis, bei der die Organe frei im Fruchtwasser schwimmen.
HÄUFIGKEIT
Beide Fehlbildungen sind insgesamt seltene Erkrankungen. Dabei kommt die Gastroschisis etwa doppelt so häufig vor wie die Omphalozele. Sie tritt bei 1 von 2‘000 bis 3‘000 Neugeborenen auf. Dabei sind Mädchen und Jungen gleich häufig betroffen. Eine Omphalozele kommt bei 1 -2 von 5´000 Neugeborenen vor, wobei Jungen etwas häufiger betroffen sind als Mädchen. Hochgerechnet auf Deutschland entspricht dies etwa 400Gastroschisis- und 200Omphalozelen-Geburten pro Jahr in unserem Land - es wird also rund ein Gastroschisis-Baby pro Tag und ein Omphalozelen Baby alle 2 Tage geboren. Die Häufigkeit der Gastroschisis ist über die letzten Jahrzehnte hinweg weltweit leicht zunehmend, die der Omphalozele ist hingegen konstant geblieben. Die Ursache hierfür ist bisher unklar.
PROGNOSE & LANGZEITERGEBNISSE
Die langfristige Prognose ist bei beiden Fehlbildungen hinsichtlich des Bauchwanddefektes äußerst günstig und in der Regel resultieren diesbezüglich keine langfristigen Einschränkungen. Nach Geburt kommt die Magen-Darm-Funktion oft verzögert in Gang. Ein ggf. vorübergehend langsameres Gedeihen wird dabei jedoch ab dem 2. Lebensjahr aufgeholt und sobald sich eine normalen Magen-Darm-Funktion eingestellt hat, ist im weiteren Verlauf mit einer vollkommen normalen Magen-Darm-Funktion zu rechnen. Einen längerfristigen Einfluss auf die Prognose können bei Omphalozelen-Patienten jedoch die bei einigen Patienten vorkommenden schwerwiegenden Begleitfehlbildungen haben. Bei Gastroschisen kommen diese nicht gehäuft vor. Auch ein seltener im Rahmen der Bauchwandfehlbildung resultierendes Kurzdarmsyndrom, bei Gastroschisen häufiger als bei Omphalozelen, kann die Lebensqualität langfristig zu einem gewissen Grad beeinflussen.
DIAGNOSE & PLANUNG
Sowohl die Gastroschisis als auch die Omphalozele werden beide in der Regel bereits in der Frühschwangerschaft (ab der 12.SSW) im Schwangerschafts-Ultraschall diagnostiziert. Die frühe Diagnose ermöglicht es, die Eltern in Ruhe zu beraten und die weitere Betreuung, Geburt und Therapie zu planen. Die weitere Betreuung sollte in einem Perinatalzentrum mit entsprechender neonatologischer und kinderchirurgischer Expertise stattfinden. In Rostock und zentralem Mecklenburg Vorpommern bietet dies beispielsweise im Perinatalzentrum Mitte (Level I) des Südstadtklinikums in Kooperation mit unserer kinderchirurgischen Klinik der Universitätsmedizin Rostock.
UNSER BEHANDLUNGSKONZEPT: BETREUUNG & THERAPIE AN DER KINDERCHIRURGISCHEN KLINIK DER UNIMEDIZIN ROSTOCK
Die Betreuung findet - wie generell bei kinderchirurgischen Krankheitsbildern Neugeborener - in enger Zusammenarbeit mit dem Perinatalzentrum (Level I) des Südstadtklinikums statt.Sobald die Diagnose gestellt ist, planen wir bereits frühzeitig in der Schwangerschaft ein erstes Kennenlerngespräch in unserer Kinderchirurgischen Klinik. Hierfür vereinbaren wir mit der Familie einen flexiblen Termin in unserer Kinderchirurgischen Ambulanz, bei dem wir das Krankheitsbild, die Therapie und den Ablauf nach Geburt besprechen. Hierbei können die werdenden Eltern auch einen ersten Einblick in unsere Klinik gewinnen. Bei Bedarf oder auf Wunsch können weitere Termine folgen. In der Regel findet zum Ende der Schwangerschaft nochmals ein Treffen statt. Da der Zeitraum der Geburt bekannt ist, können wir im Vorfeld bereits alles für die baldige Ankunft und Versorgung ihres Neugeborenen vorbereiten. Direkt nach der Geburt wird das Neugeborene neonatologisch versorgt unter Mitversorgung des Bauchwanddefektes durch uns Kinderchirurgen. Das intrauterin erfolgte Screening wird durch einen erneuten Check auf weitere bisher nicht bekannte Krankheiten oder Fehlbildungen ergänzt. Anschließend wird das Neugeborene unter intensivmedizinischer Begleitung direkt in unseren Operationssaal gebracht. Dies kann in den meisten Fällen bereits kurz - unmittelbar bis wenige Stunden - nach Geburt erfolgen. Je nach weiteren Befunden und Allgemeinzustand des Kindes ist manchmal jedoch zunächst eine Stabilisierung nötig.
Therapeutisch gibt es verschiedene Verfahren, um den Bauchdeckendefekt zu korrigieren:
PRIMÄRE REKONSTRUKTION mittels DIREKTVERSCHLUSS:
Komplette Korrektur des Defektes in einer Operation, in der Regel kurz nach Geburt. Die herausgetretenen Organe werden in den Bauch verlagert und die Bauchdecke darüber verschlossen. Meistens kann der Defekt durch eine Naht verschlossen werden, selten muss bei größerem Defekt oder sehr kleiner Bauchhöhle zusätzlich ein Patch (z.B. Goretex; biologisches Material) in die Bauchwand eingenäht werden, über den die Haut oft weiterhin mit einer Naht verschlossen werden kann.
SCHRITTWEISE VERLAGERUNG mittels Schuster-Plastik im sog. „Silo“ und sekundärem = verzögerten Bauchwandverschluss:
Die Organe werden hierbei in einen sterilen Sack, das „Silo“ gepackt und am Bett aufgehängt. Die Folie kann dabei als Schuster-Plastik in den Fasziendefekt eingenäht werden oder modifiziertem neuerem Verfahren mit einem elastischen Ring unter Faszienniveau fixiert werden. So verlagern sich die Organe der Schwerkraft folgend schrittweise und weitestgehend selbstständig in die Bauchhöhle. Dies kann durch eine vorsichtige schrittweise Verkleinerung des „Silos“ ergänzt werden. In der Regel ist so eine schonende Verlagerung innerhalb von 7 bis 10 Tagen möglich. Anschließend folgt die Operation zum Verschluss des Bauchwanddefektes.
KONSERVATIVES MANAGEMENT, sog. „paint and wait“ (bei Omphalozelenpatienten):
Dieses Verfahren beschreibt eine schrittweiser Korrektur ohne Operation, bei der der Omphalozelensack mit speziellen Mitteln bestrichen und schützenden Wundauflagen behandelt wird, sich sukzessive verkleinert und sekundär epithelialisiert („mit Haut überwächst“).
ZWEIZEITIGER VERSCHLUSS
Ist anfänglich der Verschluss eines großen Defektes nicht möglich, kann auch zunächst die Haut über den Organen verschlossen werden und in einem späteren Eingriff, nachdem sich die Bauchwand sukzessive ausdehnen konnte, die tieferen Bauchdeckenschichten (Faszienverschluss). Diverse Variationen wie z.B. zusätzliche Expander oder Bauchwandplastiken sind ebenfalls möglich.
In unserer Klinik favorisieren wir wenn immer möglich die PRIMÄRE REKONSTRUKTION bei der die Fehlbildung in einer Operation korrigiert wird. Den Eingriff führen wir in der Regel kurz nach Geburt, oft noch am Tag der Geburt durch. Dieser primäre Verschluss ist in der großen Mehrheit der Fälle (ca. 80-90%) möglich und zeigt insgesamt auch die besten Resultate (Einsetzen der Magen-Darm-Funktion, geringeres Infektions- und Sepsisrisiko, Komplikationen, Kosmetik). Natürlich verwenden wir hier unter der Haut versenkte selbstauflösende Fäden, wie es unser kinderchirurgischer hausinterner Standard ist.
Doch unter gewissen Umständen kann eine primäre Rekonstruktion nicht möglich sein: Durch die sich außerhalb entwickelnden Organe ist die Bauchhöhle des Babys oft sehr klein, es entsteht ein Missverhältnis und manchmal passen die Organe schlichtweg nicht in die kleine Bauchhöhle oder das Risiko eines zu hohen Druckes in der Bauchhöhle wäre zu groß. Die Organe dürfen nicht unter dem Risiko eines zu hohen Druckes in den Bauch verlagert werden, dies kann zur Organschädigung bis hin zum Multiorganversagen führen. Auch schwerwiegende Begleitfehlbildungen können eine Narkose- und Operationsfähigkeit des Neugeborenen und so die primäre Rekonstruktion in einigen Fällen verhindern.
Nach der Operation wird das Neugeborene auf unserer pädiatrischen Intensivstation weiterbetreut. Sobald es der Zustand des Kindes erlaubt, wird es oft für einige Tage auf unsere kinderchirurgische Station verlegt, bevor es nach Hause geht. Teilweise kann auch eine Entlassung direkt von der Intensivstation erfolgen. Natürlich wird die Mutter, sofern dies aus gynäkologischer Sicht möglich ist, mit aufgenommen und auch Papa darf jederzeit zum Kind. Während des gesamten Aufenthaltes wird ihr Kind aus einem multidisziplinären Team langjährig erfahrener Kinderchirurgen, Kinderanästhesisten, Kinder-Intensivmedizinern und Pädiatern wie auch unseren engagierten Kinder-Krankenschwestern betreut.
Nach der Entlassung aus dem Krankenhaus kann die weitere Betreuung in der Regel durch den Kinderarzt / die Kinderärztin erfolgen. Begleitend finden Nachkontrollen in unserer kinderchirurgischen Ambulanz statt. In den allermeisten Fällen ist der Verlauf hinsichtlich des korrigierten, ehemaligen Bauchdeckendefektes äußerst erfreulich und das Kind kann bald ohne Einschränkungen aus unseren Kontrollen entlassen werden.
Ösophagusatresie
Die Ösophagusatresie (Ösophagus - lateinisch Speiseröhre o. Schluckdarm; Atresie - griechisch „kein Durchgang“) ist eine angeborene Erkrankung, bei der die Speiseröhre in Folge einer Anlagestörung nicht durchgängig ist. Der Abstand zwischen den beiden Stümpfen der Speiseröhre ist dabei unterschiedlich lang. Zusätzlich besteht oft eine Verbindung zwischen der Luft- und der Speiseröhre, eine so genannte „tracheo-ösophageale Fistel“ (Trachea – griechisch Luftröhre; Fistula – lateinisch Rohr).
Bei einem Säugling, der mit einer Ösophagusatresie geboren wird, kann weder Nahrung noch Speichel vom Mund in den Magen transportiert werden. Besteht eine Verbindung von der Luftröhre zum unteren Stumpf der Speiseröhre, kann Magensaft in die Lunge gelangen. Das führt zu Atemproblemen und einer Lungenentzündung. Umgekehrt kann über diese Verbindung auch der Magen mit Atemluft aufgeblasen werden, was wiederum Beschwerden im Bauch verursacht. Bei einer Verbindung zwischen dem oberen Stumpf der Speiseröhre zur Luftröhre können ebenfalls Nahrung und Speichel in die Lunge gelangen.
In der Regel wird die Erkrankung bereits in der Schwangerschaft während der Vorsorgeuntersuchungen durch die Sonographie erkannt. Anzeichen einer Atresie kann zum Beispiel zu viel Fruchtwasser in der Amnionhöhle bei gleichzeitig nicht sichtbarer Magenblase sein. In 50% aller Fälle mit einer Atresie bestehen weitere assoziierte Fehlbildungen an der Wirbelsäule, am Anorektum, am Herz, den Nieren und/oder den Extremitäten.
Nach der Diagnose einer Ösophagusatresie folgt die operative Korrektur der Fehlbildung. Dabei hängt die Art des operativen Verfahrens vom Ausmaß der Atresie, also dem Abstand zwischen den Stumpfenden und den Verbindungen zur Trachea, ab. Bei einem kurzen Abstand können innerhalb einer Operation wenige Tage nach Geburt beide Speiseröhrenanteile direkt miteinander verbunden werden. Dieser Eingriff kann sowohl offen chirurgisch als auch minimalinvasiv über die Thorakoskopie (Brustkorbspiegelung) erfolgen. Zeigt sich unmittelbar nach der Geburt, dass der Abstand zwischen den Stumpfenden zu groß ist, wird in den ersten 48 Stunden nach Geburt über einen Bauchschnitt in Magenhöhe ein künstlicher Zugang zum Magen angelegt, über den der Säugling ernährt wird. Gegebenfalls sind weitere Operationen notwendig, um die Verbindung vorzubereiten. Erst 2-3 Monate später erfolgt dann die definitive Verbindung der beiden Stumpfenden. Das Verfahren entspricht dabei der Operation in den ersten Lebenstagen. Sollten sich die Enden vor dem Eingriff nicht ausreichend angenähert haben, kann durch Verlagerung des Magens oder Verwendung von Dünn- oder Dickdarm als Speiseröhrenersatz die Lücke geschlossen werden.
Über die unterschiedlichen Operationsmethoden und Alternativen beraten wir sie sehr gerne bei Bedarf.
Nach der Operation bleibt der Säugling für ungefähr 2 Wochen bei uns im Krankenhaus. Unmittelbar nach der Operation bleibt er für ca. 3 Tage intubiert. Danach beginnt die Ernährung über die liegende Magensonde. Bis dahin kann eine Ernährung als Infusion über die Vene (parenteral) erfolgen. Wenn nach 10-12 Tagen eine Röntgendarstellung mit Kontrastmittel eine gute Durchgängigkeit der Anastomose zeigt, erfolgt der Zug der Magensonde und der Beginn der normalen Ernährung. Für uns ist es wichtig, dass jedes Kind individuell ist und seine Zeit braucht, bis es sich an die durchgängige Speiseröhre gewöhnt hat. Sobald die Nahrungsaufnahme gut funktioniert und auch Gewicht zugenommen wird, folgt die Entlassung nach Hause.
Damit endet aber nicht die Behandlung. Im Verlauf der nächsten Jahre erfolgen regelmäßige Kontrolluntersuchungen in unserer Ambulanz. Eine wichtige Funktion spielt dabei die Elterninitiative KEKS e.v. (www.keks.org). Durch diese erhält jeder Patient ein individuelles Nachsorgebuch, in dem der Verlauf dokumentiert wird und anstehende Untersuchungen geplant werden können. Sie als Eltern finden hier Unterstützung von Personen, die bereits die gleichen Probleme bewältigen mussten.
Kinder mit operierter Ösophagusatresie haben in der Regel eine gute Lebensqualität. Die typischen Komplikationen sind wiederkehrende bronchiale Infekte, bedingt durch eine Weichheit des Luftröhrenknorpels, der klassischerweise mit der Ösophagusatresie einhergeht. Mit zunehmendem Alter nimmt diese Flexibilität des Knorpels ab, wodurch die Beschwerden zurückgehen. Eine andere Ursache für die Infekte ist ein möglicher Reflux (Zurückfließen) von Magensäure aus der Speiseröhre in die Luftröhre, der vermutlich durch einen unzureichenden Verschluss am Mageneingang entsteht.
Onkologische Chirurgie
Tumorchirurgie und supportive onkologische Chirurgie
Krebserkrankungen im Kindesalter sind selten. Wenn Ihr Kind davon betroffen ist, sind Ihre ersten Ansprechpartner der Kinderarzt und Kinderonkologe. Insgesamt braucht es zur Behandlung ein großes Team von medizinischen Disziplinen, die alle auf Kinder spezialisiert sind. Nach der Diagnosestellung und den ersten Gesprächen gilt es natürlich erst einmal den Schock zu verarbeiten, die Ängste zu lindern und auch dem Kind dem Alter entsprechend zu vermitteln, was in der nächsten Zeit passieren wird.
Bei schnellem Behandlungsbeginn bestehen bei den meisten Tumorerkrankungen ein gutes Ansprechen und entsprechend hohe Chancen auf Heilung. Die häufigsten onkologischen Diagnosen bei Kindern und Heranwachsenden betreffen das Blut- und Immunsystem der Kinder. Dazu zählen die Leukämien und Lymphome. Aber es gibt auch die soliden Tumoren. Die Tumore des Gehirn und zentralen Nervensystems werden operativ durch die Kollegen der Neurochirurgie versorgt.
Wichtigster Behandlungsschwerpunkt fast aller Tumorerkrankungen ist die Chemotherapie. Für den Beginn einer Chemotherapie benötigt ihr Kind einen dauerhaften Gefäßzugang. Dazu werden durch die Kinderchirurgie entweder implantierbare Katheter oder spezielle Kinderports implantiert, die unter der Hau verlaufen. Diese verbleiben dann bis zum Abschluss der Chemotherapie. Welches Kathetersystem zu Ihrem Kind passt, wird nach sorgfältiger Abwägung entschieden.
Ports und untertunnelte Kathetersysteme
Ein Portkatheter besteht aus einer Kammer, die als Reservoir für die verabreichten Infusionen dient, und einem daran angeschlossenen dünnen Schlauch aus Kunststoff oder Silikon. Dieser wird in eine ausreichend große Vene im Bereich der Schulter- und Halsregion eingeführt und reicht bis kurz vor den rechten Herzvorhof. Die Injektionskammer liegt mehrere Zentimeter davon entfernt geschützt unter der Haut. Werden Medikamente verabreicht, muss die Haut mit einer Portnadel durchstochen werden. Eine spezielle Silikonmembran ermöglicht dabei wiederholtes Anstechen der Kammer. Die Größe der Kammer ist variabel, sie muss aber für jeder neue Injektion neu angestochen werden. Erfolgt die Gabe mehrerer Medikamente unmittelbar nacheinander, ist keine neue Punktion notwendig
Um daher eine wiederkehrende Traumatisierung gerade bei kleinen Patienten zu vermeiden, wird meist ein Hickman- oder Broviac-Katheter eingesetzt. Auch bei der Implantation dieses Katheters wird ein großes Gefäß unter Ultraschallkontrolle punktiert. Zudem wird ein Teil des beschichteten Schlauches unter der Haut durchgeführt und mehrere Zentimeter entfernt aus der Haut ausgeleitet. In diesem untertunnelnden Bereich des Katheters befindet sich eine spezielle Muffe (Cuff), die für ein Einwachsen des Katheters sorgt und aufgrund einer speziellen Beschichtung aufsteigende Infektionen vermeidet. Das Katheterende besteht aus 2 Ausgängen, aus denen schmerzfrei Blutentnahmen vorgenommen und Infusionen verabreicht werden können.
Dadurch werden schmerzhafte Neupunktionen der Venen vermieden und das Infektionsrisiko für den immungeschwächten kleinen Patienten reduziert. Nach Abschluss der Behandlung entfernen wir den Katheter in Narkose unter sterilen Bedingungen.
Solide Tumoren
Solide Tumore des Kindesalters sind u.a. Neuroblastome, Nephroblastome (Wilms-Tumoren), Hepatoblastome oder auch Knochentumore.
Bei einigen dieser Erkrankungen wird zur Diagnosestellung eine Biopsie benötigt. In der Regel erfolgte diese in Narkose oder Sedierung. Auch dabei steht für uns die Schmerzfreiheit und die sorgfältige Aufklärung im Vordergrund.
Die vollständige Entfernung eines Tumors, sofern sie möglich ist, erfolgt je nach Stadium und Art der Erkrankung zu unterschiedlichen Zeitpunkten. Eine operative Versorgung erfolgt immer erst nach abgeschlossener Diagnostik, ggf. vorheriger Chemotherapie, wenn alle Fragen der besorgten Eltern beantwortet sind und sich ein großes Team aus allen Fachrichtungen auf die Operation vorbereitet hat, zum bestmöglichen Ergebnis für ihr Kind.
Trichterbrust
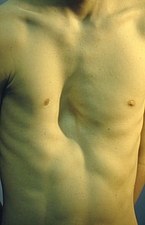

Trichterbrust und ihre operative Korrektur (MIRPE = Minimal Invasive Repair of Pectus Excavatum)
Mit einer Häufigkeit von 1: 4.00 bis 1: 1000 auftretend, stellt die Trichterbrust ein überwiegend kosmetisches Problem dar, das aber auch zu psychischen Leidensdruck und damit einhergehenden Korrekturwunsch führt. Ätiopathologisch werden genetische Faktoren als auch eine Störung des Knorpelstoff- wechsels angenommen.
Nach ausgiebiger Diagnostik zum Ausschluss organischer Begleiterkrankungen mit EKG, Echosonographie (Herzultraschall), Spirometie (Lungenfunktionsuntersuchung) sowie eines MRT´s zur Bildgebung erfolgt die Operationsplanung in einem ausführlichen Arztgespräch. Bei diesem wird die Operationstechnik nach Nuss erklärt. Die Narkose erfolgt unter Kinderanästhesiologischer Obhut.
Dabei wird unter thorakoskopischer Sicht ein individuell vorgebogener Bügel (pectus bare) in den Brustkorb unter dem eingedrückten Brustbein eingebracht und nach Umwenden der Trichter ausgedellt. Abschließend wir der Bügel mittels Halteplatten am äußeren Brustkorb fixiert. Die Operation dauert etwa 45 min. Nachfolgend wird anfänglich eine Betreuung auf der Kinder-ITS zur adäquaten Schmerztherapie vorgenommen. Nach ca. 7-10 Tagen kann umgestellt auf orale Schmerzmittel und nach erlangter Mobilisation unter physiotherapeutischer Anleitung die Entlassung in die Häuslichkeit erfolgen. Aktiv Sport kann nach ca. 3 Monaten wieder betrieben werden, davon ausgenommen ist Kampfsport. Der Bügel wird für ca. 2,5-3 Jahre belassen und sollte dann entfernt werden, um kein Fehlwachstum des Brustkorbes zu provozieren.
Kindertraumatologie
Kinderchirurgie versteht sich als Chirurgie des wachsenden Organismus. Dies ist in keinem Bereich so offensichtlich wie in der Kindertraumatologie. Der kindliche Knochen unterliegt bis zum Abschluss des Längenwachstums einem ständigen Auf- und Umbau. Seine Entwicklung verläuft über Knochenkerne im noch knorpeligen gelenkbildenden Anteil des Knochens sowie an Sehnenansätzen, über Epiphysen- und Apophysenfugen. Daraus resultieren in jeder Altersgruppe differente Röntgenbilder. Die Kenntnis des altersentsprechenden Normalbefundes ist Voraussetzung für die Diagnose einer knöchernen Verletzung. Vergleichsaufnahmen der Gegenseite sind Ausdruck mangelnder Expertise und werden von uns aus strahlenhygienischen Gründen abgelehnt.
Kinder sind durch weiteres Wachstum in der Lage, Fehlstellungen von Knochenbrüchen in einem bestimmten Rahmen auszugleichen. So können mehr als die Hälfte aller kindlichen Frakturen ohne funktionelle Einschränkungen konservativ behandelt werden. Die Entscheidung für eine konservative Therapie kann aber nur individuell im Einzelfall getroffen werden. Neben allgemeinen Faktoren, wie Alter, Geschlecht und körperliche Entwicklung spielen auch Frakturlokalisation, ihre Nähe zu einer potenten Wachstumszone, Art und Umfang der Fehlstellung und die Beziehung zur Bewegungsebene eine entscheidende Rolle.
Übersteigt das Ausmaß der Dislokation das Ziel der konservativen Frakturbehandlung, die Ausheilung in anatomiegerechter Stellung ohne funktionelle Einschränkungen, muss auch in der Kindertraumatologie eine chirurgische Intervention erfolgen. Die erforderliche Reposition in Narkose beinhaltet die definitive Versorgung mit optimaler Frakturstellung und sicherer Retention. Dabei ist eine differenzierte kindgerechte Therapie zu fordern:
- möglichst geschlossene Frakturreposition
- geringes operatives Weichteiltrauma
- möglichst frakturferne Implantation des Osteosynthesematerials
- Verhinderung von Wachstumsstörungen
- rasche Frakturkonsolidierung
- frühe Mobilisation und Belastung
- einfache Entfernung des Osteosynthesematerials
Die von uns standardmäßig verwendeten Osteosyntheseverfahren, von bewährten perkutan eingebrachten Kirschnerdrähten bis zu modernen, minimal invasiv implantierten Elastisch Stabilen Intramedullären Nägeln (ESIN) und kanülierten Schrauben, erfüllen diese Forderungen.
Bei speziellen Fragestellungen, wie z.B. arthroskopischen Gelenkoperationen oder komplexen Handverletzungen, erfolgt eine enge Zusammenarbeit mit unseren spezialisierten Kollegen der Klinik für Unfall- und Wiederherstellungschirurgie.
Wundversorgung
Wir bilden ein weites Spektrum der kindlichen Wundversorgung ab. Dieses reicht von Riss-Quetschwunden bzw. Schnittwunden über thermische Verletzungen bis hin zu Bisswunden.
Dabei legen wir ein ganz besonderes Augenmerk auf das Erzielen eines optimalen kosmetischen Ergebnisses für unsere kleinen Patienten. Das Verfahren richtet sich nach Art, Lokalisation und Ausmaß der Verletzung.
Für uns steht das Wohlbefinden unserer Patienten im Vordergrund. Für die Wundversorgung bieten wir neben der lokalen Betäubung mittels Injektion unser sogenanntes „Zauberpflaster“ an. Das „Zauberpflaster“ (Emla®-Pflaster) entspricht einem wirkstoffhaltigem Salbenpflaster, welches die Haut durch Auflage betäubt. Sollte eine Lokalanästhesie mittels Injektion durch die Haut notwendig werden, kann es zuvor auf die Einstichstelle aufgebracht werden, um den Einstichschmerz aufzuheben oder deutlich zu mindern.
Ausgedehnte Verletzungen können in Allgemeinnarkose im Operationssaal versorgt werden.
Nach Prüfung des Impfstatus wird bei Bedarf entsprechend der STIKO-Empfehlungen auf Wunsch umgehend eine Immunisierung vorgenommen.
Riss-Quetschwunden, Schnittwunden:
Die Versorgung von Riss-Quetschwunden im Bereich des behaarten Kopfes erfolgt nach Wundreinigung vornehmlich mittels Gewebekleber. Dies ist eine sehr schmerzarme Methode des nahtlosen Wundverschlusses.
Je nach Lage, Größe und Spannungsverhältnissen kommen für die Wundversorgung auch das Anlegen von Steristrips in Frage. Diese dünnen Pflasterstreifen adaptieren die Wundränder und werden bis zum Abschluss der Wundheilung belassen. Für dieses Verfahren ist, ebenso wie für die Wundrandadaptation mittels Gewebekleber, keine Betäubung durch Injektion eines Lokalanästhetikums notwendig. Eine Nahtentfernung entfällt dementsprechend ebenso.
Bei größeren, klaffenden Wunden kann eine chirurgische Naht erforderlich werden. Hierbei wird nach Wundreinigung und lokaler Betäubung genäht, um ein ansprechendes kosmetisches Ergebnis zu erzielen.
Thermische Verletzungen:
Thermische Verletzungen, insbesondere Verbrühungen, stellen ebenso eine häufige Verletzungsart im Kindesalter dar. Hierbei kommt es, je nach Tiefe der Verletzung, zur narbenlosen Ausheilung bis hin zur Notwendigkeit der Defektdeckung durch Spalthaut.
Zur primären Wundversorgung werden bei uns standardmäßig Lomatuell-Pro-Polyhexanidverbände angelegt. Das beschichtete Kontaktnetz minimiert das Risiko der Verklebung und ermöglicht damit einen schmerzarmen, atraumatischen Verbandswechsel. Zudem unterstützt es die Wundheilung. Polyhexanid ist ein zur Infektionsprophylaxe und Infektionsbehandlung angewendetes Antiseptikum. Wir verwenden es in Form einer eigens in unserem Hause hergestellten Salbe.
In Abhängigkeit von der Größe des betroffenen Areals im Verhältnis zur Körperoberfläche kann eine stationäre Aufnahme zur Infusionstherapie zum Ausgleich des Flüssigkeitsverlustes sowie eine suffiziente intravenöse Schmerztherapie erforderlich sein.
Bissverletzungen:
Auch Bissverletzungen , beispielsweise durch Hunde, Katzen o.a. Tiere, werden durch uns versorgt. Der Wundsäuberung und Spülung sowie einer möglichen adaptierenden Naht kann eine antibiotische Therapie, ggf. unter stationären Bedingungen, folgen. Insbesondere Katzenbissverletzungen kommen dabei aufgrund des Keimspektrums eine besondere Bedeutung hinsichtlich des Infektionsrisikos zu.
Bei speziellen Verletzungen des Gesichts- oder Handbereiches erfolgt eine enge Zusammenarbeit mit den Kollegen der Mund-Kiefer-Gesichtschirurgie bzw. der Handchirurgie.
Je nach Wunde kann die weitere Nachsorge durch den eigenen Haus-Kinderarzt oder bei speziellen Verletzungen oder Fragestellungen durch uns erfolgen.
Exkurs zur Stuhlinkontinenz
Die rekto-anale Kontinenz resultiert aus einem komplexen Zusammenspiel motorischer, sensorischer und anatomischer Mechanismen. Eine Inkontinenz ist die Folge einer Störung eines oder mehrerer Faktoren in diesem fein abgestimmten, komplizierten Prozess.
Wesentlich für eine Stuhlkontinenz ist neben der Stuhlhaltefunktion die Fähigkeit, Stuhl willkürlich entleeren zu können. Stuhlkontinenz ist eine unwillkürliche Fähigkeit.
Das Kontinenzorgan besteht aus dem Analkanal, dem Enddarm, dem Beckenboden und den Nervenbahnen, die das Kontinenzorgang steuern. Die Darmentleerung besteht aus einer Kombination aus Entspannung und Anspannung. Der Schließmuskel muss dabei entspannen und der Analkanal sich gleichzeitig elastisch verkürzen. Der Mastdarm muss Spannung aufbauen und sich zusammenziehen und den Stuhl nach außen drücken.
Störungen der Kontinenz können unterschiedliche Ansätze haben.
1. Unvollständiger Verschluss des Schließmuskels (Sphinkterinkontinenz)
Ist der Schließmuskelkomplex nicht in der Lage, den Analkanal dauerhaft geschlossen zu halten, ist der Anus ständig etwas geöffnet.
Kneift der Betroffene bewusst den Anus zusammen, so kann kurzfristig ein Verschluss der Analöffnung erreicht werden. Die Fähigkeit eines Patienten durch willkürliche Maximalanspannung (Zusammenkneifen des Anus) Druck im äußeren Schließmuskel aufzubauen, ist aber nicht gleichbedeutend mit einer intakten Kontinenz.
Der analgesunde Mensch verschließt seinen Darm nicht, in dem er dauerhaft willkürlich den Anus zusammenkneift. Der Schließmuskel ist in Ruhe und bei Aktivität geschlossen, ohne dass es einer bewussten Maßnahme bedarf. Als Folge des mangelhaften Verschlusses der Analöffnung kommt es zur unwillkürlichen Expression kleiner Stuhlmengen, dem Stuhlschmieren, oder einem Schleimhautprolaps als Ausdruck einer Schließmuskelinkontinenz.
2. Unvollständige Entleerung des Enddarms (Obstipation)
Die Füllung des Enddarms bewirkt das Gefühl von Stuhldrang. Wird der Entleerungsreiz ignoriert und der Darm nicht entleert, speichert der Mastdarm den Stuhl. Nach einem kurzen zeitlichen Intervall, löst der Mastdarm erneut das Gefühl des Stuhldrangs aus. Wird das Gefühl des Stuhldrangs konsequent ignoriert, kommt es zu einer Stuhlansammlung im Enddarm.
Der Darm dickt den gesammelten Stuhl ein. Die Stuhlkonsistenz wird fester. Das Stuhlkaliber nimmt zu. Der Analschleim, der normalerweise den Stuhl ummantelt, fehlt. Eine Stuhlentleerung erfordert in der Folge höheren Druck des Mastdarms und eine größere Entspannung und Aufweitung des Analkanals, um den Stuhl zu entleeren. Wird der Mastdarm nicht vollständig entleert, entwickelt sich eine Überlaufsymptomatik. Der Schließmuskel entspannt unwillkürlich, ohne dass eine Entleerung gewünscht ist. Es kommt zur unwillkürlichen Expression kleiner Stuhlmengen, dem Stuhlschmieren, als Ausdruck einer Überlaufinkontinenz.
Ziel der Therapie einer Stuhlinkontinenz ist die soziale Kontinenz des Patienten. Soziale Kontinenz bedeutet, dass der Patient mit Hilfe von Behandlungsmaßnahmen in der Lage ist, sich in seinem sozialen Umfeld ohne Einschränkung zu bewegen. Zu berücksichtigen ist, dass jede Therapie einer Stuhlinkontinenz lediglich das Symptom Stuhlentleerungsstörung und Inkontinenz behandelt, ohne die zu Grunde liegende Ursache beheben zu können.